Синдром на Марфан: признаци, диагноза, причини, симптоми и лечение
Синдромът на Марфан се проявява чрез различни патологии на скелета, сърдечно-съдовата система, нарушения на органите на зрението. Тази болест е наследствена, тя е вродена, а съединителната тъкан на тялото страда с нея.
Какво се удари на първо място
Синдромът на Марфан е нарушение на нормалното функциониране на съединителната тъкан. Тъй като се развива неправилно, то засяга много вътрешни органи. Пациентите могат да получат следните нарушения в организма:
- гигантизъм;
- аортна аневризма;
- долихостеномелия и арахнодактилия;
- късогледство;
- ектопия на лещата;
- kyphoscoliosis;
- деформация на гръдната кост;
- плоски стъпала;
- ектазия на дура матер;
- ацетабуларна издатина.
По този начин има общи физически особености при хората, които са били диагностицирани с това (синдром на Марфан). Снимките на пациентите показват сходството на външните признаци на заболяването и специалистът ще разбере доста от външния вид на пациента. Междувременно, при слаба проява на това заболяване, външните признаци могат да бъдат почти невидими. 
Как се поставя диагнозата?
Това заболяване в медицинската практика е рядко. Според експерти, само 10-20 хиляди души могат да имат синдром на Марфан. Диагностиката се основава на няколко метода. Това е фамилна анамнеза, както и проучвания на телесните функции, очен преглед, рентгенова и генетична прогноза. В същото време няма разлика какъв пол е човек, към каква раса принадлежи и на какво място се намира Земята. Това заболяване засяга в еднаква степен и жителите на юг и на север.
Ако лекарят диагностицира синдрома на Марфан, лечението може да бъде както консервативно, така и хирургично. Необходимо е да се прибягва до хирургическа интервенция в тежки случаи, когато лечението с медикаменти и физиологични процедури не дават желаните резултати.
Хирургията често е подложена на сърдечно-съдовата система и органите на зрението.
Причини за заболяването
Защо се появява синдромът на Марфан? Причините са в мутациите на гена FBN1. Той е този, който е отговорен за синтеза на фибрилин. Този структурен протеин придава на съединителната тъкан необходимата контрактилност и еластичност. При дефицит на фибрилин в тялото слабо формирани влакнести структури. Съединителна тъкан докато губи своята сила и еластичност. Много по-лошо е да издържи физиологичния стрес. От това започват да понасят вътрешни органи и скелет на човек. 
Кой рискува най-много
Както вече беше отбелязано, синдромът на Марфан, чиито признаци могат да се появят още в първите години от живота на човека, се наследява. Ако някой от семейството страда от това генетично заболяване Възможно е детето да наследи и това заболяване.
Но също така се случва, че плодът все още има основна мутация в утробата. В този случай, колкото по-възрастна е жената, толкова по-голям е рискът да има болно дете. Това е особено вярно за жени, които забременяват след 35 години.
Външни прояви
Най-често това заболяване се забелязва дори от външни признаци. Освен това, колкото по-възрастен става пациентът, толкова по-изразени са външните отличителни черти. При хора, които имат анамнеза за синдром на Марфан, снимката, както вече споменахме, е много подобна.
Скелетът им има определени особености - тялото е сравнително кратко, високо, крайниците са дълги и тънки, непропорционални на скелета. Пръстите са също продълговати, паякообразни. 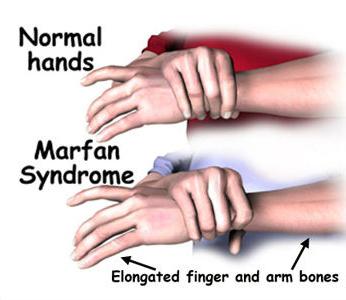
Синдромът на Марфан има симптоми като:
- астенично тяло;
- недоразвита подкожна тъкан;
- мускулна хипотония;
- тесен и дълъг лицев скелет (долихоцефалия);
- високо дъговидно небе, както и нарушение на ухапване (прогнатия).
При раждане на дете с синдром на Марфан, дължината на тялото при момчетата е над 53 см, при момичетата - 52,5 см. Крайната височина се наблюдава съответно в областта 192 и 175 см. Има и по-високи стойности.
Степента на тежест
Това заболяване има различна степен на увреждане. Съответно, хората с диагноза синдром на Марфан могат да имат различни симптоми.
Лекарите разпределят няколко различни форми на това заболяване, в зависимост от това какви са системите на живот.
Първата форма се изтрива. Промените и аномалиите в развитието са леки. Само една или две системи на тялото страдат, а не много.
Изявена е втората форма на синдрома на Мафран. Тук има няколко опции:
- Промените са леки, но присъстват в трите системи на тялото.
- В една система има очевидни отклонения от нормата.
- Промените са ясно изразени в две, три или повече системи.
Лекарите също отбелязват три тежест на заболяването: лека, умерена и тежка. Курсът на заболяването може да бъде различен. Налице е стабилен синдром на Марфан, чиито признаци през годините на наблюдения остават непроменени. Налице е прогресиращ тип заболяване, когато с течение на времето патологията на развитието расте и се влошава.
Някои особености на заболяването
Както вече отбелязахме, синдромът на Марфан се характеризира с:
- комбинирани увреждания на скелета, очите, сърдечно-съдовата и нервната системи;
- разнообразие от физиологични прояви;
- първите признаци на заболяването могат да бъдат диагностицирани както при раждане, така и след години;
- хронично прогресивно течение.
При това заболяване се наблюдава устойчива дисфункция на ставите. Експертите наричат това състояние хипермобилност, когато ставите лесно се извиват в различни посоки и много мобилни.
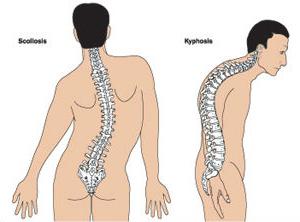
Много пациенти, страдащи от синдрома на Марфан, имат неправилна структура на гръдния кош, придобива форма на фуния или кил.
Ортопедите често отбелязват деформация на гръбначния стълб в различните му прояви:
- сколиоза;
- kyphoscoliosis;
- кифоза;
- изкълчвания и сублуксации на цервикалния регион;
- спондилолистези.
Пациентите често страдат от плоска стъпалост и издатина на ацетабулума.
Сърдечни усложнения
Сърдечно-съдовата патология често доминира в клиничната картина на това заболяване. Тя се проявява по различни начини. Пациентът може да има постоянни дефекти в стените на кръвоносните съдове. Те губят своята еластичност. Особено чувствителни към това са аортата, големите клони на белодробната артерия. Пациентите могат да получат малформации на клапния апарат и сърдечните стени.
Що се отнася до аортата, най-често се наблюдава прогресивно разширяване на неговата възходяща част и клапанния пръстен (дилатация, анулоартозна ектазия). Пациентът често има аневризми, митралната клапа е засегната. Има и патологично удължаване на хордите и тяхното разкъсване.
При дете, което все още е в утробата, но на генно ниво, синдромът на Марфан вече е бил предаден на него, често се образуват вродени сърдечни дефекти. Те могат да бъдат:
- коарктация на аортата ;
- дефект на предсърдната преграда (ASD);
- белодробна стеноза;
- дефект на интервентрикуларната преграда (VSD).
Могат да възникнат и нарушения на сърдечния ритъм (тахикардия, предсърдно мъждене), развитие на инфекциозен ендокардит.
В най-неблагоприятната неонатална форма на синдрома на Марфан, сърдечната недостатъчност прогресира бързо от самото раждане на детето. Често тези деца не живеят до една възраст.
Нарушена визуална функция
Пациентите с това заболяване в повечето случаи страдат от патология на органа на зрението. Те могат да бъдат:
- късогледство;
- изравняване и увеличаване на размера на роговицата;
- дислокация / сублуксация на лещата;
- хипоплазия на ириса и цилиарния мускул;
- промяна в калибъра на ретинаталните съдове;
- кривогледство.

В същото време, на възраст от 4 години, ектопията на лещата често се развива, тя напредва бързо, зрителната функция се влошава.
Основно лечение
Заслужава да се отбележи, че при това заболяване един лекар не може да предпише лечение. Експерти от различен профил извършват наблюдение и препоръки: офталмолог, ортопед, кардиолог, кардиохирург, терапевт, генетик.
На първо място, лечението има за цел да предотврати прогресирането на това заболяване. Тъй като накрая е невъзможно да се отървете от болестта, важно е да го спрете, за да не се засегнат жизнените органи в бъдеще.

Ако има провал на клапите на сърцето, тогава се показва операция. С неотложна нужда да се постави протезна митрална клапа.
Бременните жени, чийто синдром на Марфан има изразена кардиоваскуларна патология, се отнасят за ранно цезарово сечение.
Ако зрението е нарушено, специалистите избират специални очила и контактни лещи. При очевидни патологии, когато се правят диагнози като катаракта, глаукома, изместване на лещата, се предписва хирургично или лазерно лечение.
Също така операцията се показва, ако има изразени нарушения в скелетната система. Лекарите държат спинална стабилизация, торакопластика, артропластика на бедрото.
Колко пациенти живеят с това заболяване
При синдрома на Марфан вероятността от внезапна смърт е висока. Само специалистите могат да предскажат колко дълго човек ще живее с такава болест, като се има предвид степента на увреждане на тялото. Навременното лечение и корекцията на сърдечната хирургия могат да подобрят качеството на живот и да увеличат продължителността му до 60-70 години. Не трябва да се забравя, че хората със синдром на Марфан трябва да се подложат на диагноза и да бъдат постоянно наблюдавани от лекар.
Лекарите ограничават физическата активност при пациенти с това заболяване, те са противопоказани за високи натоварвания, гмуркане, контактни спортове.
Жени, които имат анамнеза за това заболяване и планират да имат деца, трябва да бъдат прегледани от генетик.